
- 什么是FDA 510 k许可?
- 类型的510 ks
- 在哪里开始美国FDA 510 k审批程序。
- 谁需要FDA 510 k,为什么?
- FDA 510 k的设备标识点
- FDA 510 k医疗器械审批流程
- FDA 510 k申请准备
- 什么是验收评审?
- 什么是实质性审查?
- FDA批准了这个词到底是什么意思?
- FDA不批准什么?
- FDA采用基于风险的分级方法管理医疗器械。
还阅读:
- FDA医疗器械审批流程分步指南
- 美国食品药品监督管理局医疗器械广告和促销条例
- DMF的类型以及FDA对DMF Type III的清晰度的重要性
- 美国食品和药物管理局发布关于在大流行期间注册和进口设备的常见问题
- FDA检查与ISO审核:FDA 21 CFR Part 820和ISO 13485
- 了解FDA 21 CFR part 820与iso 13485之间区别的重要性
- FDA批准的510(k)s医疗器械收益和风险分析最终指南
- 医疗器械制造商的FDA QSR符合性最终指南
- FDA 510(k)医疗设备的Premarket通知|超声波透热疗效
- FDA公布改善医疗器械安全的行动计划
- 510k Vs上市前批准
什么是FDA 510 k许可?
希望在美国市场贸易的每个设备制造商,为分类医疗设备I,II和III。如果这些意味着人类使用,没有必要,预先达尔批准申请(PMA)是不必要的。But must submit FDA 510 k unless the device is exempt from 510 k necessity of the Federal Food, Drug, and Cosmetic Act (the FD&C Act), and does not overreach the circumstance of exemptions of the device classification regulation chapters (e.g., 21 CFR 862.9, 21 CFR 864.9).
FDA 510k是由FDA制定的上市前批准流程,表明即将上市的器械至少与未获上市前批准的合法上市器械(21 CFR 807.92)一样安全有效,实质上等同。申请人必须将其医疗器械测量为一种或多种合法销售的类似器械,以支持其实质等同主张。值得注意的是,FDA不执行510 k预许可设施检查。申请人可以在510 k许可后立即销售他们的设备在FDA 510 K清除后的任何时间。
FDA不“批准”FDA 510 k提交。“清除”。宣传一款通过510(k)认证的设备为“fda批准”是不合法的。(阅读更多)
- 510 K清关
《食品、药品和化妆品法》第510(k)条要求必须注册的器械制造商至少提前90天通知FDA其销售医疗器械的意图。这被称为上市前通知-也称为PMN或510(k)。
- 美国FDA 510k认证


谁需要FDA 510 k,为什么?
国内医疗器械来自任何国家的制造商试图向美国市场推出一个新设备。重新打包或重新标签,使标签更改或其操作显着影响设备。简单的单词,FDA 510 K是一个过程的名称,但技术上,提交您的设备文档进行清除的复杂和同样重要的过程,以确定设备是否安全有效,因此允许在美国市场合法销售。.

FDA 510 k的设备标识点
仅当与谓词相比,一个设备仅被认为是相同的
- 设备必须具有与谓词相同的预期用途;和
- 此外,设备应该具有与谓词相同的技术特征;
- 故意使用应该与谓词保持一致
- 该装置是否具有不同的技术特性,并没有提出不同的安全性和有效性问题
- 提交给FDA的数据显示,该设备至少与合法的市场设备一样安全有效。
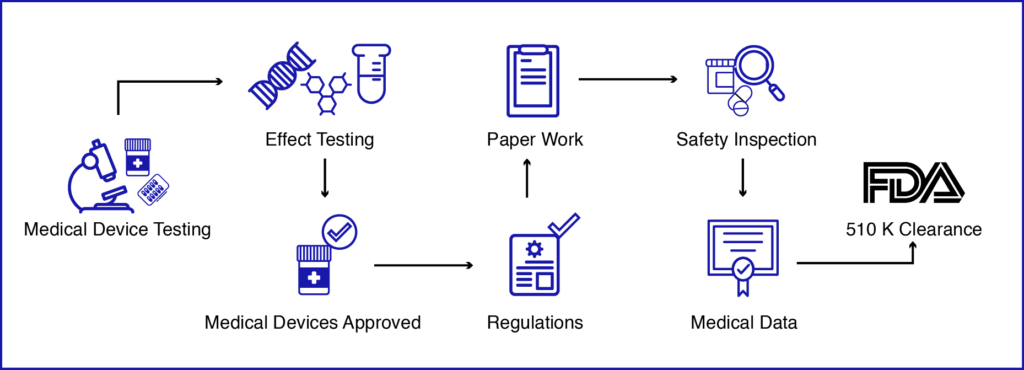
FDA 510 k医疗器械审批流程
相当等价的状态并不意味着新的和谓词装置必须相同。在预期用途、设计、使用或交付的能源、材料、化学成分、生产工艺、性能、安全性、有效性、标签、生物相容性、标准和其他适用特征方面建立显著等效性。外国制造商/出口商或外国制造商/出口商的美国代表向美国市场介绍设备。
FDA 510 k申请准备
任何想在美国销售设备的人都需要在设备销售前至少90天向美国FDA提交510万,即使在此日期之前它可能已经在开发或临床研究中。改变你已经在商业发行的设备的预期用途。如果一种合法销售的器械发生了变化或修改,而这种变化可能会显著影响其安全性或有效性。对于现有器械的更改或修改,需要提交新的美国FDA 510k文件,这些修改可能会显著影响该器械的安全性或有效性,或者该器械将用于新的或不同的适应症。使产品GMP不豁免的医疗器械制造商应实施21 CFR 820部分作为质量管理体系。
符合21 CFR第820部分质量管理体系要求的GMP非豁免产品。美国FDA 510万美元的申报应与审查费用一起准备和提交,根据营业额可以显著降低美国FDA 510万美元的审查费用。包括子公司在内的实体营业额在1亿美元以下才有资格被认定为SBU (Small Business Unit)。该审查由设备和辐射健康中心进行,如有疑问,将提出质疑。成功完成活动后,美国FDA批准510 k编号。在美国FDA 510 k批准后,为了在美国供应产品,工厂注册和器械上市必须完成。
什么是验收评审?
一旦发出确认函,将DCC路线提交给适当的ODE或OIR部门,首席审稿人进行验收评审
- 使用验收清单评估510(k)在管理上是否完整
验收评审的结果通常是:
- 510(k)被接受实质性审查;或
- 该510(k)未被接受审查(即认为拒绝接受或RTA);或
- 由于FDA没有在15个日历天内完成验收审查,510(k)正在进行实质性审查。
提交人有180个日历天来完全解决在等举行.如果不这样做,510(k)将被视为撤回并从我们的审查系统中删除。如果510(k)被删除,510(k)提交人将需要提交一份新的、完整的510(k),以获得FDA对该设备的上市许可。
188金宝搏网站靠谱吗博彩战略家是FDA 510 K流程顾问帮助客户注册SBU(小型企业单位),如适用。取出产品的测试要求,创建档案,解决疑问和完成所有活动后,客户收到美国FDA510 k上市前批准.我们还协助建立注册和器械清单,使医疗器械在美国合适的供应。
什么是实质性审查?
在实质性的审查,引导审稿人对510(k)提交的全面审查综合审查,并通过a与提交者进行沟通实质性互动,应在收到510(k)提交材料后60个日历日内提交。
的实质性互动通信通常是:
- 一封电子邮件,指出FDA将继续解决任何未偿还的缺陷互动评论;或
- 电子邮件
- 电话
- 请注意:在互动评论,提交给DCC的任何信息必须包括一份有效的副本。
- 一个额外的信息(ai)请求暂停提交的请求。
- 提交人有180个日历日,从日期人工智能的请求提交一份完整的回复人工智能的请求.
FDA批准了这个词到底是什么意思?
FDA负责通过控制人类药物和生物制品、动物药物、医疗设备、烟草物品、食品(包括生物食品)、化妆品和辐射电子物品来保护公众健康。
在任何情况下,没有那么多的产品通过上市前批准——也就是说,在产品上市前,由FDA专家和办公室批准的安全性和充分性审计。食品药品监督管理局的要求不时地集中在产品现在可以购买之后。
FDA不批准什么?
FDA不批准公司。FDA不“批准”卫生保健设施、实验室或制造商。FDA确实有权检查受监管的设施,以验证它们符合适用的良好生产规范规定。国内或国外食品、药品和大多数器械设施的所有者和管理人员应向FDA登记他们的办公室,除非适用排除。血液和组织办公室也应与该办公室合作。
FDA采用基于风险的分级方法管理医疗器械。
FDA根据风险对设备进行分类。最高风险的设备(III类),如机械心脏瓣膜和可植入的输液泵,通常需要FDA在营销前批准预售批准应用。为了获得这些设备的FDA批准,制造商必须具有足够的,有效的科学证据,即可以合理保证这些设备对其预期用途安全有效。
一般来说,一旦证明中等风险的医疗器械(II类)(例如透析设备和许多类型的导管)实质上等同于不需要上市前批准的合法销售的断言设备,FDA就会“批准”其上市。对使用者造成伤害的风险较低的设备(I类)(例如无动力吸奶器、弹性绷带、压舌器和检查手套)仅受一般控制,大多数免于上市前通知要求。
今天联系我们为优秀服务和负担得起的咨询费
